

The latest Public Health England (PHE) Technical Briefing on the variants of concern is out, and it shows that the infectiousness of the Delta (Indian) variant has dropped again, so that it is now at the same level as the Alpha (British) variant was at the end of March.
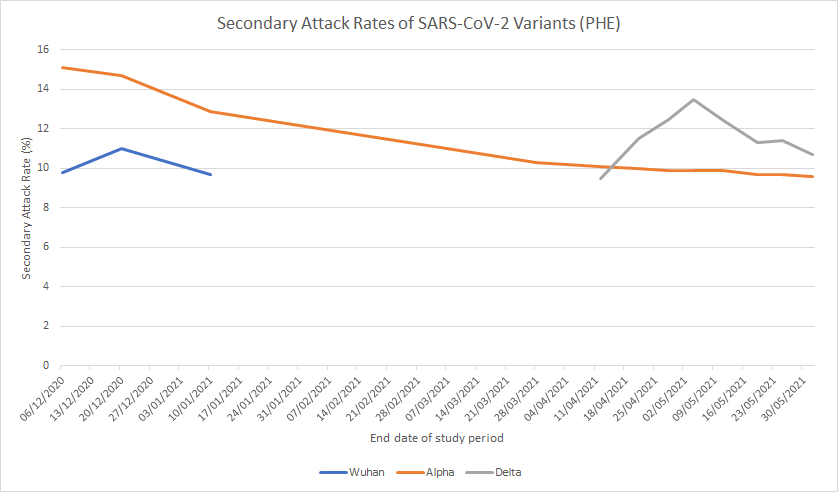
The Delta variant secondary attack rate (SAR – the proportion of contacts an infected person infects) is down to 10.7% this week, from 11.4% a week ago (see graph above). That means that around 90% of the close contacts of people infected with the Delta variant are not infected by them. Once again, this shows how claims that the Delta variant is “60% more transmissible” or similar fail to make clear that transmissibility changes and often declines over time, and that even if the Delta variant is currently more infectious than the Alpha variant it is still less infectious than the Alpha variant was when it first appeared on the scene.
You might be wondering, though, how a disease that only infects 10-15% of close contacts, even at the height of an epidemic wave, can be called highly infectious. And indeed, how such a disease can cause an explosive epidemic wave at all, and why those waves crash down as quickly as they arise despite not exhausting the pool of susceptible people.
If you do wonder that, you would not be alone. A famed epidemiologist called Robert Edgar Hope-Simpson spent his life observing the idiosyncrasies of seasonal influenza and summarised some of his thinking in a 1981 article entitled “The role of season in the epidemiology of influenza“. More recently, in 2008, a group of scientists took up Hope-Simpson’s investigations (which were inconclusive) and summarised seven conundrums of influenza that he identified, all of which apply equally to COVID-19:
1. Why is influenza both seasonal and ubiquitous and where is the virus between epidemics?
2. Why are the epidemics so explosive?
3. Why do epidemics end so abruptly?
4. What explains the frequent coincidental timing of epidemics in countries of similar latitudes?
5. Why is the serial interval [the time it takes for secondary cases to appear] obscure?
6. Why is the secondary attack rate so low?
7. Why did epidemics in previous ages spread so rapidly, despite the lack of modern transport?
The scientists add two more:
8. Why does experimental inoculation [infection] of seronegative [without antibodies] humans fail to cause illness in all the volunteers?
9. Why has influenza mortality of the aged not declined as their vaccination rates increased?
To explain these strange features, Hope-Simpson proposed the radical notion that, unlike other infectious diseases like measles and non-seasonal cold-causing viruses like rhinovirus, influenza is not primarily spread by transmission from the sick to the well (hence the low SAR). Rather it is spread by temporarily highly infectious and usually symptomless carriers in whom the virus has been latent but has now been triggered by a seasonal stimulus. Hope-Simpson was never able to identify the seasonal stimulus, though argued it must be ultimately dependent on variation in solar radiation, which is the driver of the seasons.
The scientists behind the 2008 article are not persuaded by the idea of latency of influenza, which they suggest has no evidence. Instead, they think the behaviour can be explained by seasonal variations in innate immunity, especially caused by the drop in vitamin D during winter months:
Significant seasonal and population variations in innate immunity make it unnecessary to postulate latency to explain the bizarre epidemiology of influenza… To make sense of influenza’s epidemiology, we revise Hope-Simpson theory, hypothesising marked variation in the infectivity of the infected (the good infectors demonstrated in rats by Schulman and Kilbourne in 1963) and that vitamin D deficiency is Hope-Simpson’s seasonal stimulus.
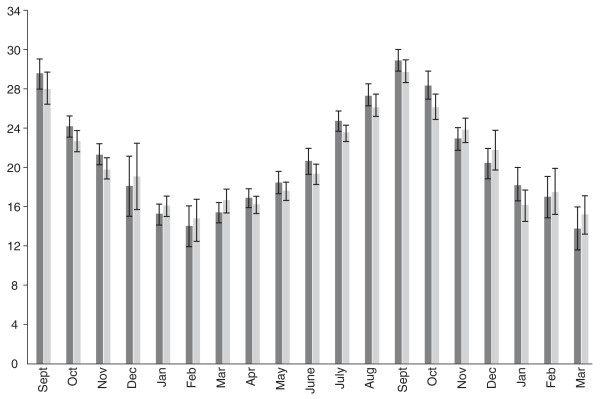
This theory leaves questions of its own, however, such as why a gently undulating vitamin D level would suddenly trigger a brief explosion in viral infectivity, and why the flu season typically starts in August and September, when vitamin D is at its peak (see figure below).
Recently there has been a lot of interest in the idea of “super spreaders“. This is the counterintuitive idea that with diseases like flu and COVID-19 the majority of infected people don’t pass on the virus at all and a small number (say 10-20%) are responsible for almost all transmission. In fact, this is now a widely accepted concept of epidemiology – which suggests Hope-Simpson was onto something.
The question remains though: what triggers the brief but deadly explosive outbreaks of seasonal viruses like flu and COVID-19, and what brings them just as abruptly to an end? Hope-Simpson’s observation, that it must have something to do with changes in solar radiation, since it is a seasonal phenomenon which occurs simultaneously in geographically remote places of the same latitude, remains pertinent.
The disappearance of flu in the last 18 months adds to the mystery (and ideas that flu has simply been re-badged as COVID-19 don’t stack up against the huge amount of genetic sequencing of variants that has been done).
Whatever the underlying cause of the strange behaviour, though, some facts are clear. SARS-CoV-2 is not, by any reasonable definition, a highly transmissible virus – infected people on average only infect 10-15% of their close contacts, even at the height of the winter surge (and that is an upper bound as it assumes that all subsequent infections were caused by the index case rather than being co-index cases). This compares to 57% for rhinovirus and 70% for measles – those are highly transmissible viruses. Rather, COVID-19 is subject to the same kind of mysterious and explosive seasonal triggering as influenza is. This probably helps to explain why lockdowns make no clear impact on transmission or mortality, and why the virus does not immediately rebound when restrictions are lifted, as all the models keep wrongly telling us is going to happen.
One lesson from this is we should stop imagining we can capture the behaviour of this strange disease in simple mathematical models and pay more attention to real-world data. Perhaps then we would see things much more in proportion, and cease redesigning our world around our exaggerated fear of it.









To join in with the discussion please make a donation to The Daily Sceptic.
Profanity and abuse will be removed and may lead to a permanent ban.